

The challenge of new biosimilar drugs
2014/11/01 Calvo Hernáez, Begoña - Farmazian eta Farmazia Teknologian katedradunaFarmazia Faktultatea. EHU Iturria: Elhuyar aldizkaria
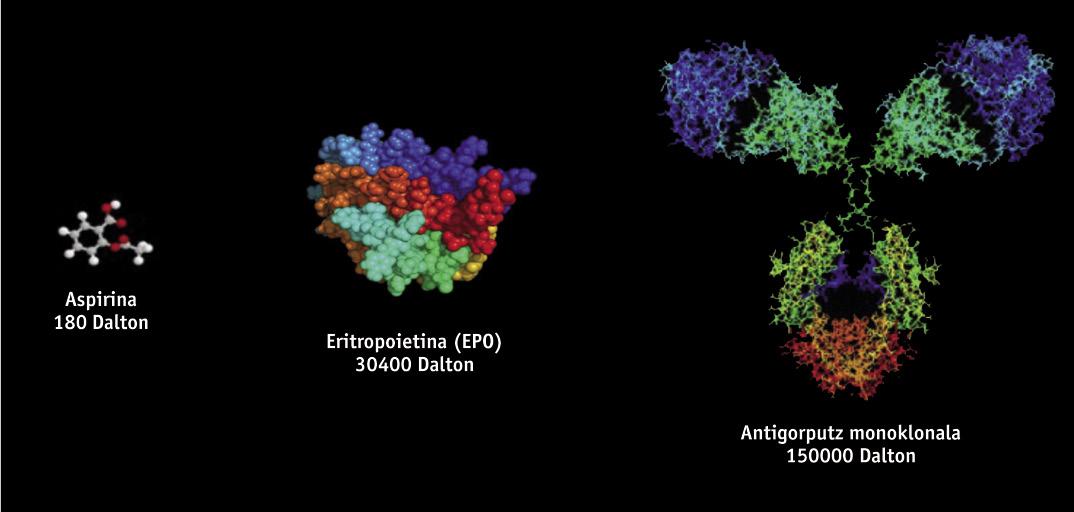
Biotechnological drugs, of protein structure, are obtained through genetic engineering techniques, such as recombinant DNA, so it is very difficult to study and characterize them, unlike generic drugs obtained by chemical synthesis. Biotechnological drugs are obtained from genetically modified living organisms such as bacteria, yeasts and certain cell lines of animal and plant origin. Since different cell lines and production processes are used in manufacturing, there are no two products of completely equal biological origin. Therefore, since copies of original drugs are similar but not exactly the same, generic regulations are not scientifically suitable for biosimilars. Small variations of the molecule or small modifications in the manufacturing process can affect the result and the final product, so its manufacture needs a specific regulation.
It is estimated that biosimilar drugs are 20% cheaper than original products, obtaining less savings than generic ones, so they are 40% cheaper than original ones. However, although the savings rate is lower because they are used in expensive therapies, their introduction into clinical practice will be a great advantage for the healthcare system and will allow patients to access costly therapies.
On the other hand, the entry of biosimilars into the market represents an increase in competition, which will logically result in a decrease in the price of this type of products. In this way, health budgets will be more sustainable and resources such as support for health research lines may be allocated for other purposes.
Biosimilars in Europe
Biosimilar drugs approved so far in Europe include products such as growth hormone, erythropoietin (EPO) or interferon. Recently, the first biosimilars of monoclonal antibodies have been approved, such as infliximaba, which is used for the treatment of autoimmune diseases (rheumatological, psoriatic diseases, Crohn's diseases, among others). Monoclonal antibodies are even more complex molecules than initial biosimilars, and presumably they will have an "enormous influence." It is estimated that until 2020 twenty billion euros could be saved using biosimilar monoclonal antibodies.
In Spain, the implantation of admitted biosimilars is not the same in all cases: some biosimilars have achieved a 50% implantation (such as filgrastima, which is used to combat the decrease in defense of cancer patients treated with chemotherapy), others have achieved a lower implantation: for example, erythropoietin have achieved an implantation of approximately 20% and growth hormones of the order of 5%.
Biosimilars are accepted in our environment through a centralized procedure in Europe. The European Medicines Agency (EMA) is responsible for defining the requirements that these products must meet for marketing in the European Union. Following the publication in 2004 of the legal framework on biosimilars by the SEM, a series of guidelines and standards have been published on quality requirements, both general and product-specific, as well as preclinical and clinical trials prior to product approval. The EMA has been a pioneer in the world in establishing such regulations for the authorization of biosimilars. In fact, other regions and developed countries have relied on European regulations to publish their regulations: Australia, Japan, etc. The same applies to the guidelines established by the World Health Organization (WHO) and the U.S. Food and Drug Agency (the latter is in the process of approving the first instructions on biosimilars).
Finally, the appearance of these copies of biological drugs, as well as the appearance of generics, has generated a debate about the possibilities of guaranteeing the same quality, efficacy and safety as the drugs of origin. Doctors who prescribe this type of drug should know that biosimilars do not get authorization if they are not shown to be comparable to the original products. To do this, the SEM establishes strict controls and requirements. Therefore, it is essential that the health personnel who prescribe and use this type of drug have scientific, precise and solid information on the peculiarities of these treatments.

Gai honi buruzko eduki gehiago
Elhuyarrek garatutako teknologia

