

El reto de los nuevos medicamentos biosimilares
2014/11/01 Calvo Hernáez, Begoña - Farmazian eta Farmazia Teknologian katedradunaFarmazia Faktultatea. EHU Iturria: Elhuyar aldizkaria
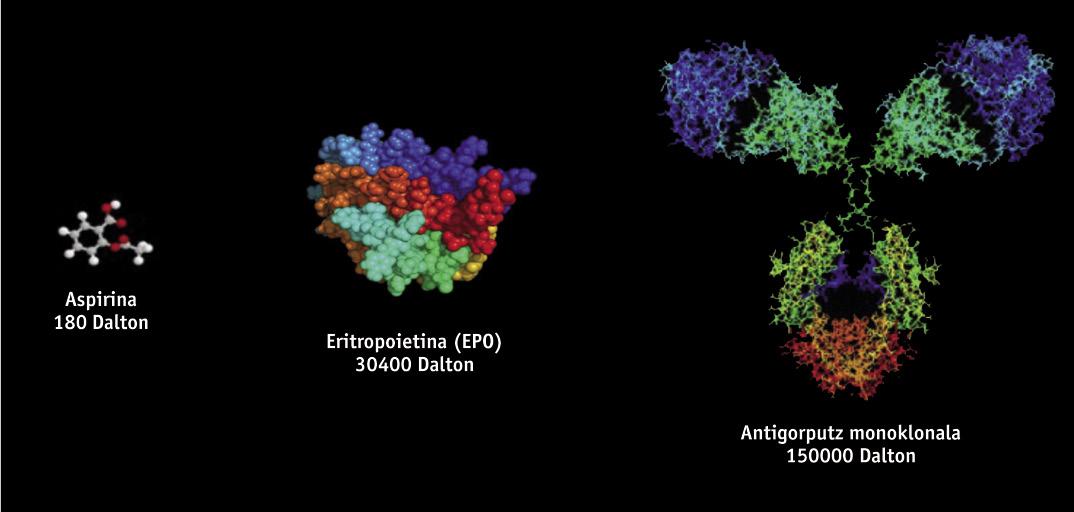
Los medicamentos biotecnológicos, de estructura proteica, se obtienen mediante técnicas de ingeniería genética, como el ADN recombinante, por lo que es muy difícil su estudio y caracterización, a diferencia de los medicamentos genéricos que se obtienen mediante síntesis química. Los medicamentos biotecnológicos se obtienen a partir de organismos vivos modificados genéticamente, como bacterias, levaduras y ciertas líneas celulares de origen animal y vegetal. Dado que en la fabricación se utilizan diferentes líneas celulares y procesos productivos, no existen dos productos de origen biológico totalmente iguales. Por ello, dado que las copias de medicamentos originales son similares pero no exactamente iguales, la normativa de genéricos no es científicamente adecuada para los biosimilares. Pequeñas variaciones de la molécula o pequeñas modificaciones en el proceso de fabricación pueden afectar al resultado y al producto final, por lo que su fabricación necesita de una normativa específica.
Se estima que los medicamentos biosimilares son un 20% más baratos que los productos originales, obteniéndose un menor ahorro que los genéricos, por lo que son un 40% más baratos que los originales. No obstante, a pesar de que el porcentaje de ahorro es menor, debido a que se utilizan en terapias costosas, su introducción en la práctica clínica supondrá una gran ventaja para el sistema sanitario y permitirá a los pacientes acceder a terapias costosas.
Por otra parte, la entrada de los biosimilares en el mercado supone un aumento de la competencia, lo que lógicamente redundará en una disminución del precio de este tipo de productos. De esta forma, los presupuestos sanitarios serán más sostenibles y se podrán destinar recursos como el apoyo a líneas de investigación sanitaria para otros fines.
Biosimilares en Europa
Entre los medicamentos biosimilares aprobados hasta el momento en Europa se encuentran productos como la hormona del crecimiento, la eritropoyetina (EPO) o la interferona. Recientemente, además, se han aprobado los primeros biosimilares de anticuerpos monoclonales, como la infliximaba, que se utiliza para el tratamiento de enfermedades autoinmunes (enfermedades reumatológicas, psoriásicas, enfermedades de Crohn, entre otras). Los anticuerpos monoclonales son moléculas aún más complejas que los biosimilares iniciales, y es de suponer que tendrán una "enorme influencia". Se estima que hasta el año 2020 se podrían ahorrar veinte mil millones de euros utilizando anticuerpos monoclonales biosimilares.
En España, la implantación de biosimilares admitidos no es la misma en todos los casos: algunas biosimilares han conseguido una implantación del 50% (como la filgrastima, que se utiliza para combatir la disminución de la defensa de los pacientes oncológicos tratados con quimioterapia), otras han conseguido una implantación menor: por ejemplo, las eritropoyetinas han conseguido una implantación del 20% aproximadamente y las hormonas del crecimiento del orden del 5%.
Los biosimilares se aceptan en nuestro entorno a través de un procedimiento centralizado en Europa. La Agencia Europea de Medicamentos (EMA, por sus siglas en inglés) es la encargada de definir los requisitos que deben cumplir estos productos para su comercialización en la Unión Europea. Tras la publicación en 2004 del marco legal sobre biosimilares por parte de la SEM, se han editado una serie de guías y normas sobre requisitos de calidad, tanto generales como específicos para cada producto, así como de ensayos preclínicos y clínicos previos a la aprobación de los productos. La EMA ha sido pionera en el mundo en establecer este tipo de normativas para la autorización de biosimilares. De hecho, otras regiones y países desarrollados se han basado en la normativa europea para publicar sus reglamentos: Australia, Japón, etc. Lo mismo ocurre con las directrices marcadas por la Organización Mundial de la Salud (OMS) y la Agencia de Alimentos y Medicamentos de Estados Unidos (esta última organización está en fase de aprobación de las primeras instrucciones sobre biosimilares).
Por último, la aparición de estas copias de medicamentos biológicos, al igual que la aparición de los genéricos, ha generado un debate en torno a las posibilidades de garantizar la misma calidad, eficacia y seguridad que los medicamentos de origen. Los médicos encargados de prescribir este tipo de fármacos deben saber que los biosimilares no consiguen autorización si no se demuestra que son comparables a los productos originales. Para ello, la SEM establece estrictos controles y requerimientos. Por lo tanto, es fundamental que el personal sanitario que prescribe y utiliza este tipo de fármacos disponga de información científica, precisa y sólida sobre las peculiaridades de estos tratamientos.

Gai honi buruzko eduki gehiago
Elhuyarrek garatutako teknologia

