

Le défi des nouveaux médicaments biosimilaires
2014/11/01 Calvo Hernáez, Begoña - Farmazian eta Farmazia Teknologian katedradunaFarmazia Faktultatea. EHU Iturria: Elhuyar aldizkaria
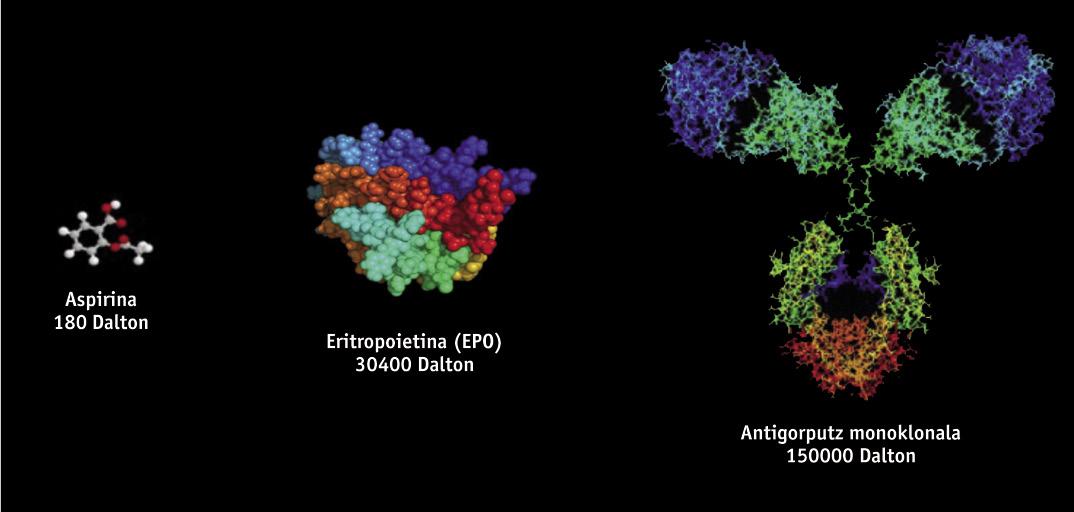
Les médicaments biotechnologiques, de structure protéique, sont obtenus par des techniques d'ingénierie génétique, comme l'ADN recombinant, ce qui rend très difficile leur étude et caractérisation, contrairement aux médicaments génériques obtenus par synthèse chimique. Les médicaments biotechnologiques sont obtenus à partir d'organismes vivants génétiquement modifiés, tels que les bactéries, les levures et certaines lignes cellulaires d'origine animale et végétale. Puisque dans la fabrication on utilise différentes lignes cellulaires et processus productifs, il n'y a pas deux produits d'origine biologique totalement égaux. C'est pourquoi, puisque les copies de médicaments originaux sont similaires mais pas exactement identiques, la réglementation des génériques n'est pas scientifiquement adaptée aux biosimilaires. De petites variations de la molécule ou de petites modifications dans le processus de fabrication peuvent affecter le résultat et le produit final, de sorte que sa fabrication a besoin d'une réglementation spécifique.
On estime que les médicaments biosimilaires sont 20% moins chers que les produits originaux, ce qui permet d'obtenir moins d'économies que les médicaments génériques, ce qui fait 40% moins chers que les produits originaux. Cependant, bien que le pourcentage d'épargne soit inférieur, étant donné qu'ils sont utilisés dans des thérapies coûteuses, leur introduction dans la pratique clinique constituera un grand avantage pour le système de santé et permettra aux patients d'accéder à des thérapies coûteuses.
D'autre part, l'entrée des biosimilaires sur le marché suppose une augmentation de la concurrence, ce qui va logiquement conduire à une baisse du prix de ce type de produits. De cette façon, les budgets sanitaires seront plus durables et des ressources telles que l'appui aux lignes de recherche sanitaire pourront être allouées à d'autres fins.
Biosimilaires en Europe
Parmi les médicaments biosimilaires approuvés jusqu'à présent en Europe, on trouve des produits tels que l'hormone de croissance, l'érythropoïétine (EPO) ou l'interférone. Récemment, en outre, les premiers biosimilaires d'anticorps monoclonaux, tels que l'infliximaba, ont été approuvés pour le traitement des maladies auto-immunes (maladies rhumatologiques, psoriasiques, maladies de Crohn, entre autres). Les anticorps monoclonaux sont des molécules encore plus complexes que les biosimilaires initiaux, et il est à supposer qu'ils auront une "énorme influence". On estime que jusqu'en 2020 on pourrait économiser vingt milliards d'euros en utilisant des anticorps monoclonaux biosimilaires.
En Espagne, l'implantation de biosimilaires admis n'est pas la même dans tous les cas: certaines biosimilaires ont obtenu une implantation de 50% (comme la filgrastime, utilisée pour combattre la diminution de la défense des patients oncologiques traités par chimiothérapie), d'autres ont obtenu une implantation mineure: par exemple, les érythropoïétines ont obtenu une implantation d'environ 20% et les hormones de croissance.
Les biosimilaires sont acceptés dans notre environnement par une procédure centralisée en Europe. L'Agence européenne des médicaments (EMA) est chargée de définir les conditions requises pour la commercialisation de ces produits dans l'Union européenne. Après la publication en 2004 du cadre légal sur les biosimilaires par la SEM, une série de guides et de normes sur les exigences de qualité, tant générales que spécifiques pour chaque produit, ainsi que des essais cliniques et précliniques ont été édités avant l'approbation des produits. L'EMA a été un pionnier mondial dans l'établissement de ces normes pour l'autorisation de biosimilaires. En effet, d'autres régions et pays développés se sont fondés sur la réglementation européenne pour publier leurs règlements : Australie, Japon, etc. Il en va de même pour les directives établies par l'Organisation mondiale de la santé (OMS) et l'Agence américaine des aliments et des médicaments (cette dernière organisation est en phase d'approbation des premières instructions sur les biosimilaires).
Enfin, l'apparition de ces copies de médicaments biologiques, comme l'apparition des génériques, a généré un débat sur les possibilités d'assurer la même qualité, efficacité et sécurité que les médicaments d'origine. Les médecins chargés de prescrire ce type de médicaments doivent savoir que les biosimilaires ne sont pas autorisés si elles ne sont pas prouvées comparables aux produits originaux. Pour ce faire, la SEM établit des contrôles stricts et des exigences. Il est donc essentiel que le personnel de santé qui prescrit et utilise ce type de médicaments dispose d'informations scientifiques, précises et solides sur les particularités de ces traitements.

Gai honi buruzko eduki gehiago
Elhuyarrek garatutako teknologia

