

Reciclar proteínas, llave para estar sano
2020/03/01 Osinalde Moraleja, Nerea - EHUko Biokimika eta Biologia Molekularra Saileko irakaslea | Elu Arantzamendi, Nagore - EHUko ikertzailea Iturria: Elhuyar aldizkaria
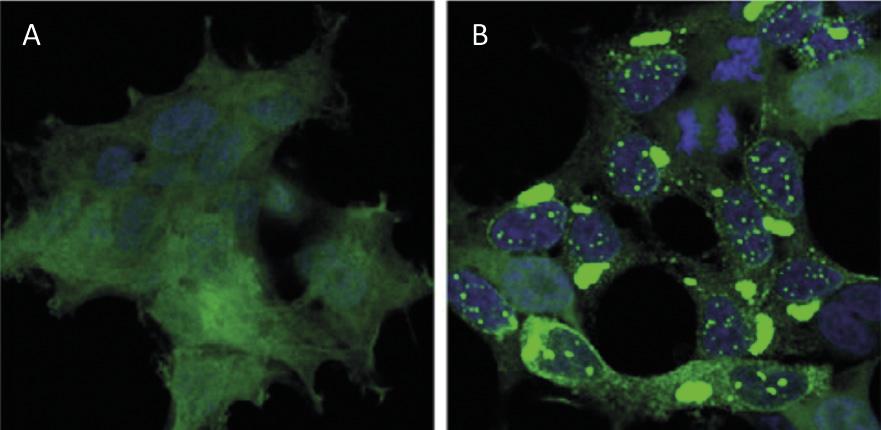
El reciclaje de proteínas a nivel celular tiene un impacto similar al del reciclaje de residuos a nivel mundial. Las proteínas son las principales moléculas que garantizan el funcionamiento de las células. Son responsables de llevar a cabo reacciones químicas, regular los genes, transportar moléculas y mantener la estructura celular. Cada célula contiene miles de proteínas que además se forman de forma muy dinámica. De hecho, para responder a las necesidades de la célula actual o a los estímulos externos, se está produciendo y destruyendo constantemente proteínas de diferentes tipos celulares. Por tanto, es imprescindible mantener el equilibrio entre estos dos procesos adversos para garantizar el correcto funcionamiento de la célula. Por ello, no es de extrañar que muchos de los procesos de síntesis y degradación de proteínas que azotan nuestra sociedad estén en el origen de muchas enfermedades. Entre las enfermedades desarrolladas como el Alzheimer, el Parkinson y el Huntington, todas ellas muy conocidas [1,2] (Figura 1).
Proteínas: bien plegadas o acabadas.
La información para la producción de proteínas está almacenada en los genes. En la cadena de ADN se especifica cuándo y qué proteína se generará. La estructura de una proteína que se extrae de la fábrica de producción de proteínas, es decir, de los ribosomas, es equivalente a un collar de perlas con aminoácidos. Sin embargo, esta proteína, para ser funcional, debe tener una determinada estructura tridimensional. En la célula se pueden encontrar 20 tipos de aminoácidos y la secuencia de aminoácidos de cada proteína determina la estructura funcional tridimensional, es decir, la estructura nativa de esta proteína. Los requisitos celulares son muy especiales, por lo que las proteínas necesitan a menudo la ayuda de proteínas denominadas chaperones moleculares para obtener una estructura nativa. Los seres humanos tenemos más de cien chapperones, cada uno de los cuales se encarga del proceso de plegado de cientos de proteínas ituales. Cuando la actividad de los txaperones está dañada por las mutaciones, el proceso de plegado de las proteínas diana no es el adecuado, por lo que estas proteínas no son funcionales. Si no se degradan, se acumulan en la célula causando graves daños [4] (Figura 2).
Por ejemplo, las mutaciones ocurridas en el txaperón denominado HSPB1 provocan una esclerosis lateral amiotrófica y una neuropatía grave denominada Charcot Marie Tooth [5,6]. Y los que tienen el chaperón CHIP mutado desarrollan el síndrome de Gordon Holmes, un mal único que causa hipogonadismo y problemas neurológicos.
Pero en todos los casos en los que las proteínas se doblan mal y se acumulan patológicamente, la culpa no es de los txaperos. En algunos casos, debido a las mutaciones que se producen en el genoma, las proteínas se producen con más aminoácidos de los necesarios, lo que impide obtener la estructura nativa correspondiente (Figura 2).
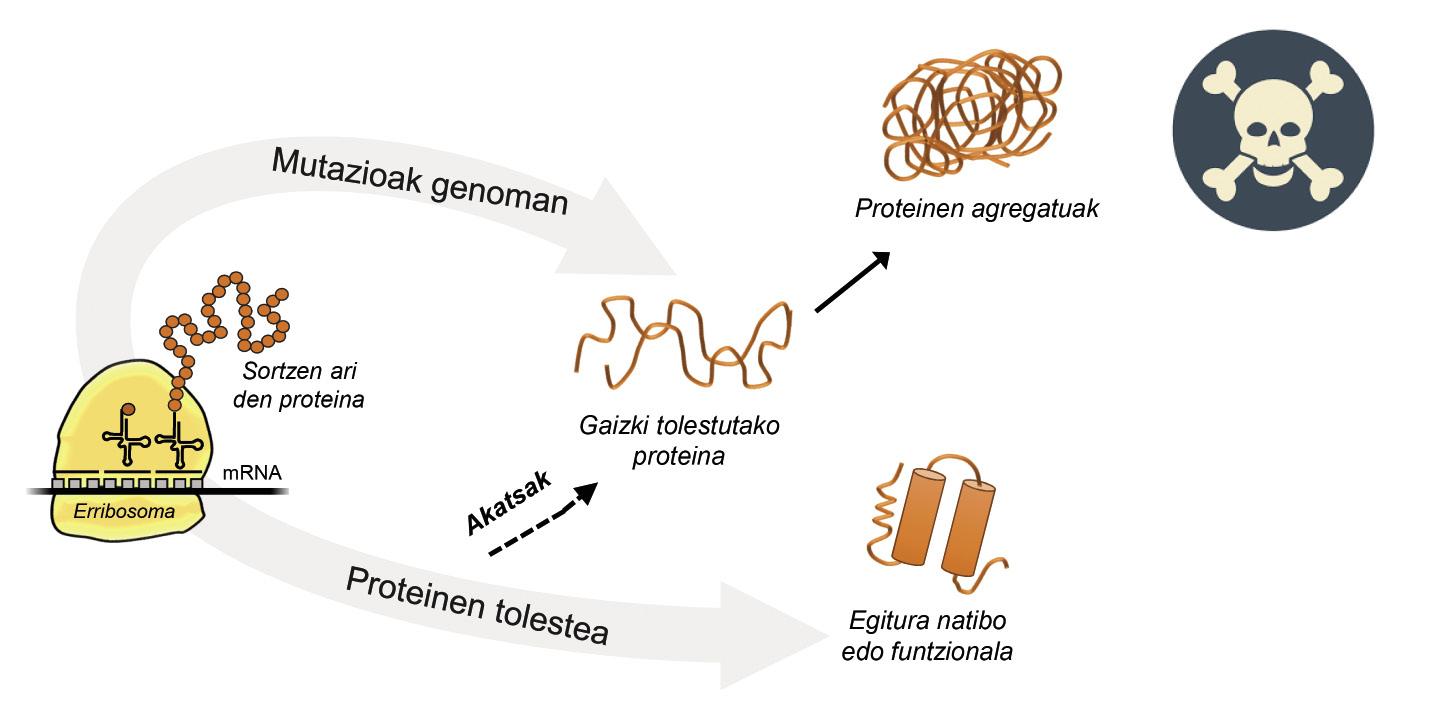
Por ejemplo, los afectados por la enfermedad de huntington tienen una proteína de huntingtina defectuosa. De hecho, al mutarse el gen HTT que codifica la huntingtina, la proteína producida contiene más aminoácidos glutamina de los necesarios, es decir, contiene demasiados ejemplares de una perla determinada [4]. Los afectados por la esclerosis lateral múltiple y la demencia frontotemporal también presentan una proteína C9orf72 defectuosa con más pares de aminoácidos glicina de los necesarios, que tiene una gran tendencia a acumularse en neuronas [7].
Sin embargo, la enfermedad más conocida por la acumulación de proteínas es el alzheimer, una enfermedad que agita fuertemente la sociedad actual. A medida que mueren las neuronas del hipocampo cerebral, las personas enfermas pierden sus capacidades cognitivas, especialmente la memoria a corto plazo. Pero, ¿por qué mueren las neuronas? Cada vez hay más evidencia de que las acumulaciones de proteínas tau y beta-amiloide mutadas son las principales responsables de la muerte de las neuronas [6].
Proteasoma: trituradora de proteínas
¿Y la célula no tiene mecanismos para degradar proteínas mal plegadas y evitar agregados patológicos? Sí, y más de una. Sin embargo, por un lado, teniendo en cuenta los ejemplos anteriormente mencionados, es evidente que algunas acumulaciones de proteínas son capaces de romper con estos mecanismos, y por otro lado, la propia máquina de degradación de proteínas puede tener problemas e impedir el reciclado.
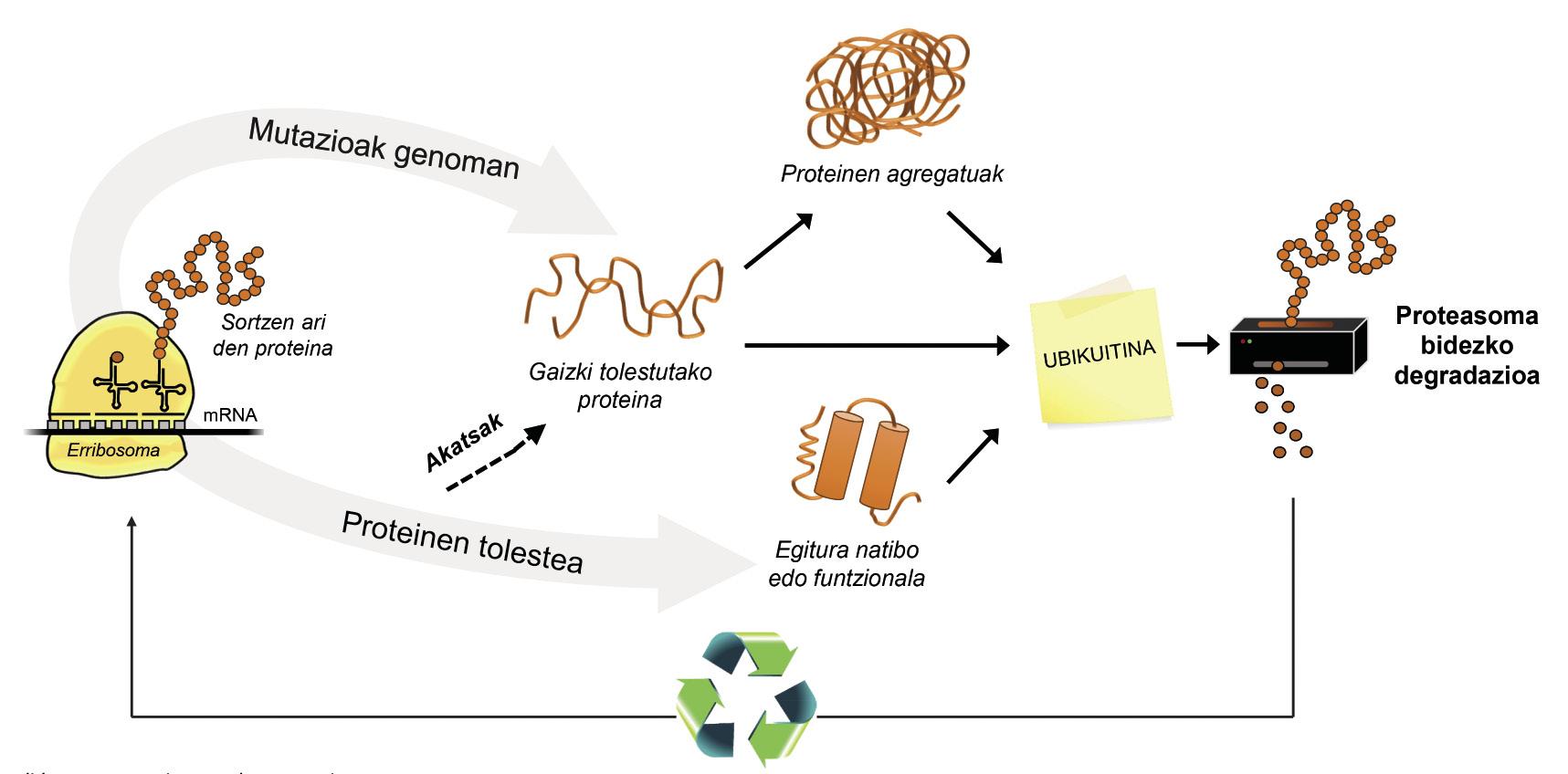
Además de las proteínas mal plegadas, la mayoría de las proteínas no disponibles para la célula se degradan gracias a un complejo multiproteico llamado proteasoma. Pero entre los miles de proteínas que hay en la célula, ¿cómo se puede saber cuál es lo que hay que degradar? ¿Cómo percibe el proteasoma qué proteínas hay que reducir? Pues porque tienen adherida una pequeña molécula llamada ubiquitina. Esta molécula funciona como un post-it que dice “debes ir a proteasoma a degradar”. Proteasoma actúa como una trituradora de papel. Reduce las proteínas, es decir, rompe las relaciones entre los aminoácidos. Paralelamente, pone a disposición de la célula aminoácidos libres para la producción de nuevas moléculas [8]. Por ello, se puede afirmar que el proteasoma recicla proteínas (Figura 3).
El proceso de reciclaje de proteínas es muy complejo y basta con que se produzca un error en cualquier punto para que se desarrolle una enfermedad. En el proceso de unión de la ubiquitina a las proteínas que van a ser transferidas a proteasoma, colaboran tres tipos de enzimas: Enzimas E1, E2 y E3. Si por estar mutadas estas enzimas el proceso de ubiquitinación de las proteínas no es correcto, las proteínas a degradar no se destruyen y se acumulan en la célula. Esta toxicidad se manifiesta sobre todo en neuronas altamente vulnerables, lo que significa que muchas enfermedades neurológicas están relacionadas con los defectos que se producen en estas enzimas [8].
La enzima UBA1 tipo E1, por ejemplo, está relacionada con una enfermedad neuromuscular excepcional que sólo afecta a los chicos. Estos pacientes, además de tener una gran debilidad muscular, no tienen reflejos, es decir, los músculos no responden adecuadamente a estímulos externos. Los que tienen mutada la enzima UBE2A tipo E2 desarrollan el síndrome de deficiencia UBE2A, una enfermedad neurológica atípica. Esta enfermedad es también padecida únicamente por hombres, siendo los síntomas más frecuentes la deficiencia mental, alteraciones cutáneas, del aparato reproductor y urinario y la ausencia de habla.
Entre las tres enzimas que realizan la ubiquitinación de proteínas, las más abundantes son las E3. Así, existe alguna enzima E3 mutada detrás de la mayoría de las enfermedades neurológicas desarrolladas como consecuencia de una ubiquitinación defectuosa. Por ejemplo, quienes sufren el autismo familiar y el autismo esporádico, clasificados dentro de los trastornos del espectro del autismo, tienen mutadas las enzimas UBE3B y UBE3C tipo E3, respectivamente. Por el contrario, las personas con UBE3A defectuosa desarrollan el síndrome de Angelman relacionado con el autismo [9].
Además de las enfermedades relacionadas con el autismo, los E3 mutados pueden causar otras enfermedades neurológicas muy diferentes. Por ejemplo, quienes tienen mutada la enzima E3 llamada gigaxonina desarrollan la neuropatía de los axones gigantes. En condiciones saludables, la gigaxonina estabiliza los neurofilamentos de los brazos neuronales, es decir, de los axones, condicionando su estructura y tamaño. Sin embargo, cuando la gigaxonina se muta, se convierte en no funcional y no vascuitina los neurofilamentos de los axones. Esto hace que no se degraden los neurofilamentos y que los axones de las neuronas sean más grandes y largos de lo necesario. En consecuencia, la mayoría de los pacientes que sufren esta neuropatía tienen problemas para caminar y, en general, para coordinar los movimientos [9].
Tras el reciente síndrome de Tenorio se encuentra una enzima E3, en este caso denominada RNF125. Los afectados por el síndrome de Tenorio tienen discapacidad intelectual. Además, este tipo de pacientes se caracterizan por un crecimiento excesivo, especialmente el desarrollo excesivo del cráneo. Lo contrario les ocurre a los que tienen mutado el TRIM36, a los que les falta gran parte del cráneo y del cerebro [9].

Conclusiones
Es evidente que un desequilibrio en el ciclo de síntesis y degradación de proteínas puede producir efectos realmente nocivos para las células y los organismos vivos en general. Por un lado, proteínas mal dobladas por mutaciones, acumuladas como bolsas de basura inutilizables, pueden causar la muerte de las células. Por otra parte, el deterioro de los dispositivos moleculares responsables de la degradación y el reciclado de proteínas produce acumulaciones tóxicas de proteínas. De forma análoga a la acumulación de basura en caso de que la actividad de una instalación de reciclaje de residuos se anulara.
Sabemos que uno de los principales retos de la sociedad actual es la correcta gestión de los residuos. Y es que, si no se garantiza el reciclaje de los residuos, en pocos años cubriremos nuestros océanos y campos con basura y la vida se hará insostenible. En el mundo microscópico las células tienen un reto similar cuando tienen problemas para gestionar proteínas no funcionales. Para garantizar su salud es imprescindible eliminar las proteínas tóxicas.
Por lo tanto, si la próxima vez se te olvida reciclar los residuos de tu hogar, recuerda el desastre que ello supondría para una célula. De hecho, el reciclaje de la basura que generamos los seres humanos y el reciclaje de las proteínas que se acumulan en las células son acciones similares, a gran y pequeña escala. Una llave para que todos podamos mantenernos sanos.
Bibliografía
